Antimicrobial removal on piglets promotes health and higher bacterial diversity in the nasal microbiota
The view on antimicrobials has dramatically changed due to the increased knowledge on the importance of microbiota composition in different body parts. Antimicrobials can no longer be considered only beneficial, but also potentially deleterious for favourable bacterial populations. Still, the use of metaphylactic antimicrobial treatment at early stages of life is a practice in use in porcine production. Many reports have shown that antibiotics can critically affect the gut microbiota, however the effect of perinatal antimicrobial treatment on the nasal microbiota has not been explored yet. To gain insights on the potential changes in nasal microbial composition due to antimicrobial treatments, piglets from two different farms were sampled at weaning. The nasal microbiota was analysed when antimicrobial treatment was used early in life, and later, when no antimicrobial treatment was used during the lactation period. Removal of perinatal antimicrobials resulted in an increased bacterial diversity in nasal microbiota at weaning. Concurrently, elimination of antimicrobials produced an increase in the relative abundance of Prevotella and Lactobacillus, and a decrease in Moraxella and Bergeyella. These changes in microbiota composition were accompanied by an improvement of the piglets' health and a higher productivity in the nursery phase.
The use of antimicrobials to control bacterial diseases is a common practice in porcine production. Despite the undeniable fact that usage of antimicrobials plays a vital role in the production of food-animals and in protection of public health 1 , they have proved to negatively affect the beneficial microbiota 2 . The microbiota is a key factor for the well function and homeostasis of different body systems in animals. Key functions of the microbiota include the correct development of the immune system and resistance against pathogens colonization or patho-genicity reduction 3–5 . The interest on animal microbiota has increased in recent years due to its impact in health. Knowledge on microbiota composition can result in the identification of potential bacterial groups associated with health 5–7 , although many factors can interfere in the establishment of a so-called adequate microbiota, such as environment, pig production system, pig genetics and antimicrobial treatments 8 . Microbial dysbiosis, which may appear secondarily to antimicrobial use, can facilitate pathogen infections and enhance the tissue damage inflicted by pathogenic bacteria 9 . The use of metaphylactic antimicrobials, especially early in life, can have a deleterious impact in animal health through the alteration of the gastrointestinal tract (GIT) microbiota compo-sition 10 . Many efforts have been made to elucidate the GIT microbiota composition and the relationship of dys-biosis with many diseases, however, there is scarce knowledge on the nasal microbiota composition in animals. In pigs, it has been demonstrated that the microbial communities inhabiting the nasal cavities at weaning may influence the development of Glässer's disease later in life 6 and perinatal antimicrobials are sometimes used to reduce the risk of this disease. However, whether the antibiotic treatment early in life has an effect on the bacterial communities from the piglet's nose, has not been explored yet. Here, we analysed the nasal microbiota compo-sition of 3–4 week old piglets at weaning regarding antimicrobial administration in the lactation phase, with the aim of detecting the effect on the microbiota compositions and the association with health status later in life.
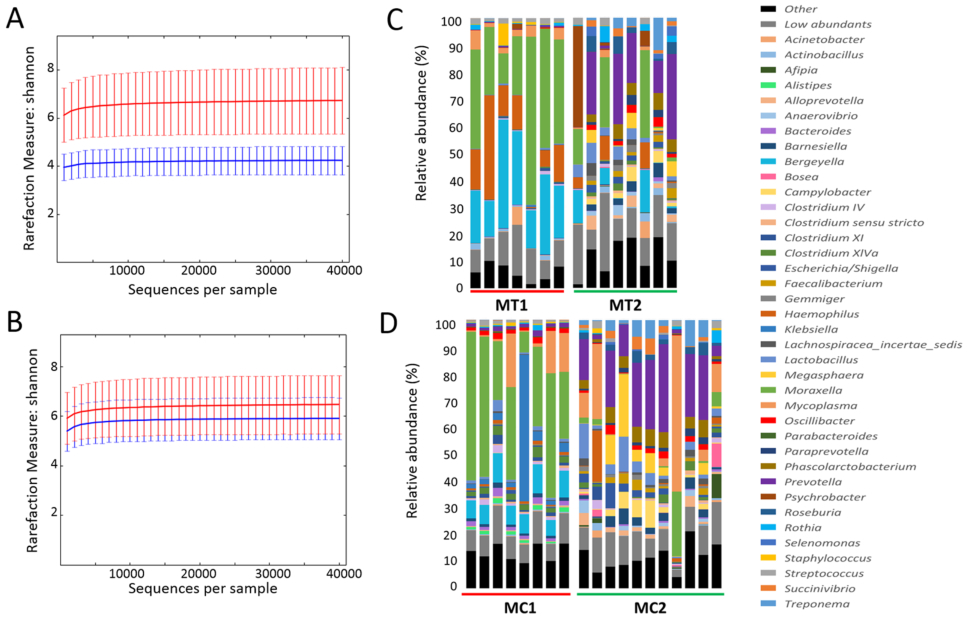
Figure 1. Nasal microbiota of piglets from farms MT and MC before (1) and after (2) antimicrobial treatment removal. Microbiota rarefaction curve generated using Shannon diversity estimator with samples from farms MT (A) and MC (B). Samples have been rarefied at an even depth of 40,000 sequences per sample. Error bars indicate the 95% confidence intervals. The mean relative abundance (%) of OTUs found in nasal swabs of piglets from farms MT (C) and MC (D) is presented at genera level.
Results
Diversity and species richness from the nasal microbiota. With the aim of analysing the effect of perinatal antibiotic treatment on the nasal microbiota composition, 3–4 week old piglets from two farms (MT and MC farms) from Spain were sampled. Nasal swabs were taken when both farms were using perinatal antimicrobial treatments (MC1 and MT1), and total DNA was extracted and subjected to individual massive sequencing. The same sampling was repeated one productive cycle after elimination of perinatal antibiotics in both farms (MC2 and MT2). We obtained a total of 8,883,783 joint reads after filtering for both farms (MC1: 876,010; MC2: 3,178,022; MT1: 2,533,426; MT2: 2,296,325). The OTUs were identified in the samples that passed the quality-based filters (MT1, n = 7; MT2, n = 8; MC1, n = 8; MC2, n = 11) through clustering sequences at 97% sequence homology. At phylum level, 98.2% of the sequences were assigned to 7 phyla, while 93% of the sequences were assigned at family level, to 41 families. At genus level, 75.5% of the reads were assigned to 68 genera. The relative abundance of the OTUs found at genus level in the nasal microbiota is represented in Fig. 1 (for the full list of OTUs assigned at gen-era level, please refer to Additional File 1; see also Additional File 2 for other taxonomical levels).
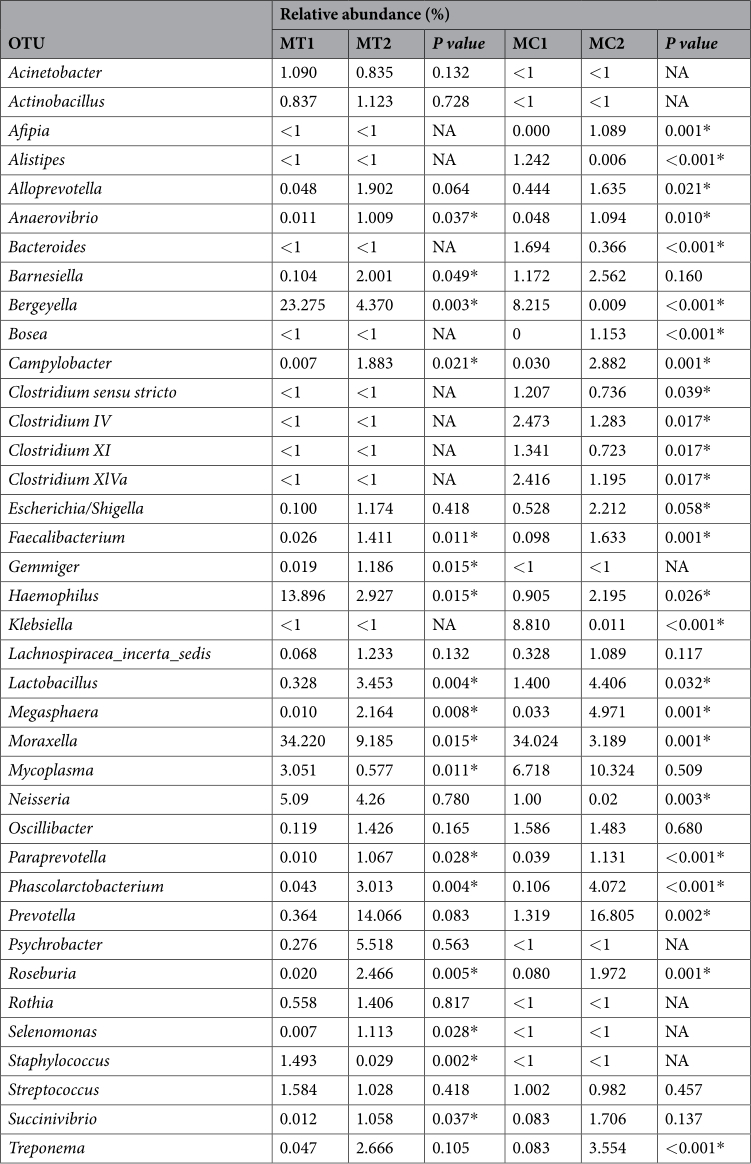
One cycle after elimination of perinatal antimicrobials, nasal microbiota of all the weaning piglets analysed showed a significant increase in bacterial diversity, as indicated by the analysis of alpha diversity through Shannon diversity index (P = 0.002). When farms MC and MT were analysed individually (Fig. 1), this increase in alpha diversity was also observed, showing to be statistically significant through non-parametric two-sample t-test (999 permutations, P ≤ 0.05) only in farm MT (Fig. 1A,B). The mean of observed OTUs richness was also measured in rarefied samples at maximum depth (40,000) at the two sampling times in both farms, with means values of 1,465 (MC1) and 2,237 (MC2) OTUs for MC farm, and 1,221 (MT1) and 4,619 (MT2) OTUs for MT farm. The most predominant bacterial genera across all the samples at first sampling time was Moraxella for both farms (34.22 ± 6.30% for MT1 and 34.02 ± 5.79% for MC1, Fig. 1C,D) followed by Bergeyella (23.27 ± 3.6% for MT1 and 8.21 ± 0.7% for MC1, Fig. 1C,D), while the most abundant genera after antimicrobial elimination in both farms changed to Prevotella (14.06 ± 4.46% for MT2 and 16.81 ± 3.26% for MC2).
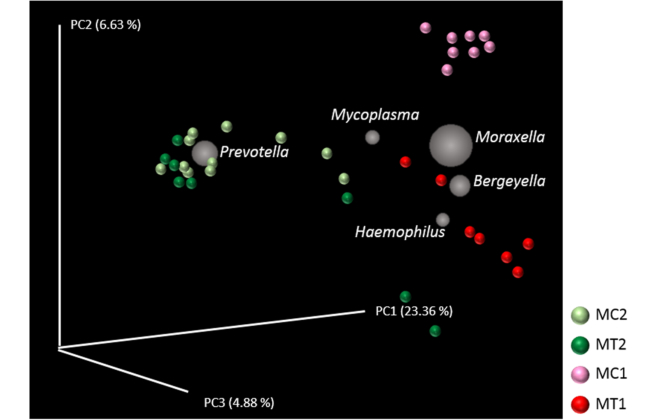
Analysis of the beta diversity both before and after elimination of antimicrobials, showed remarkable differences in the bacterial population of the nasal microbiota (Fig. 2). Antimicrobial treatment explained 22.63% of the differences observed (P = 0.001). The five most abundant genera explaining the divergence between the nasal microbiota before and after elimination of perinatal antimicrobials were: Prevotella, Moraxella, Bergeyella, Haemophilus and Mycoplasma (Fig. 2). When the beta diversity was analysed in each farm individually, significant differences were observed after elimination of the antimicrobial treatment in both farms (adonis, R 2 = 0.2186, P = 0.001 for MC and R 2 = 0.1935, P = 0.005 for MT). Elimination of antimicrobials caused significant changes in the relative abundance of several genera. Among others, Lactobacillus, Phascolarctobacterium, Megasphaera, Roseburia and Campylobacter were increased in both farms. The mean of the relative abundance of the Prevotella genus increased also after elimination of the antimicrobials, but this increase was only statistically significant for MC farm (Table 1). On the contrary, the relative abundance of Moraxella and Bergeyella significantly decreased, while Haemophilus and Mycoplasma showed a decreased tendency when antimicrobials were removed in farm MT, but increased in farm MC (Table 1). Finally, the relative abundance of Neisseria decreased after removal of antimicrobials, showing to be significant in MC farm (Table 1).
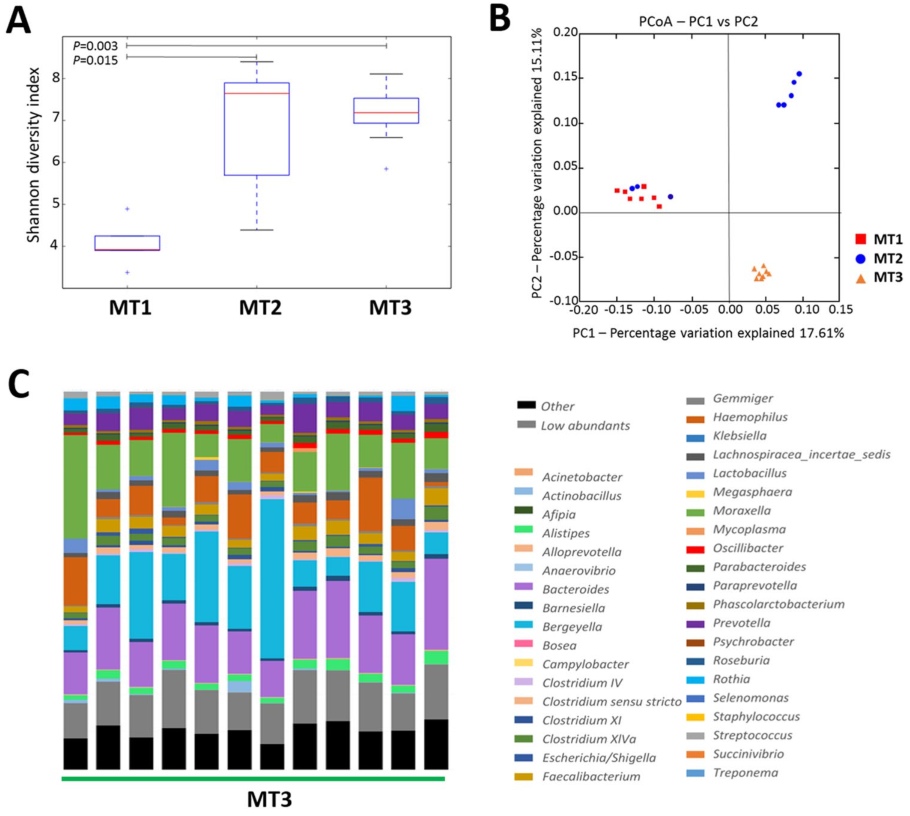
Stability of microbiota composition after antimicrobial elimination. In order to evaluate the stability of these changes in the nasal microbiota composition, a third sampling was performed in farm MT (MT3). In this run, we obtained a total of 4,003,077 joint reads after filtering (MT3, n = 12). Analysis of alpha diversity showed no further increase in diversity after additional time without perinatal antimicrobials in the farm (Fig. 3A, for a full list of OTUs assigned at genera level, please see Additional File 3). However, the samples after 5 cycles without antimicrobials showed a more similar composition demonstrated with the lower P value obtained through the comparison (Fig. 3A; Shannon index, P = 0.003). Besides, the beta diversity analysis, showed a distinct clustering at the three sampling times tested (Fig. 3B, adonis R 2 = 0.15643, P < 0.001). Sixteen genera were involved in the long-term changes observed after elimination of the antimicrobials (Table 2 and Fig. 3C). Nine of these genera were increased at the third sampling point after 5 productive cycles without antimicrobials, such as Faecaliabacterium and Bacteroides.
Health and production parameters in the nursery phase. Concomitantly to the increase in microbiota diversity, the MT farm experienced an improvement of health and productivity. Analysis of production parameters in the nursery phase from 2015 to 2017, indicated that the elimination of antimicrobial treatments resulted in a significant reduction in medication cost by pig and mortality rate, while the reduction observed in feed conversion ratio (FCR) was not statistically significant (P = 0.110) (Table 3). In farm MC, the FCR (P = 0.261) did not show a clear improvement, but the mortality rate (P = 0.130) and especially the cost in medication per pig (P = 0.05) decreased during the study period.
Discussion
The balance in the composition of the microbiota community needs to be also considered. Thus, while the abundance of some pathogenic bacterial genera decreased, the opposite was observed for some of the genera linked to beneficial effects on animal health. Besides the well-known probiotic genus Lactobacillus, other potential beneficial genera were detected in this study, such as Prevotella and Bacteroides. Prevotella has been reported previously as a beneficial and/or protective bacteria or at least an important component of healthy microbiota of animals and humans 27,28 . Although some Bacteroides have been considered opportunist pathogens 29–31 , at least one strain within the species Bacteroides fragilis has been identified as a potential probiotic for human health 32 . In general, an in-depth analysis of the specific species and strains is needed to define the specific role of each taxon within each genus and its potential role in animal health.
The present study confirms the benefits of the elimination of perinatal antimicrobial usage in the health of piglets, through modulation of the nasal microbiota composition. Based on the long-term results presented herein, antimicrobial treatments should be carefully applied especially at early stages in life, when the cross-talk with commensal microorganisms will determine the microbiota establishment throughout life.
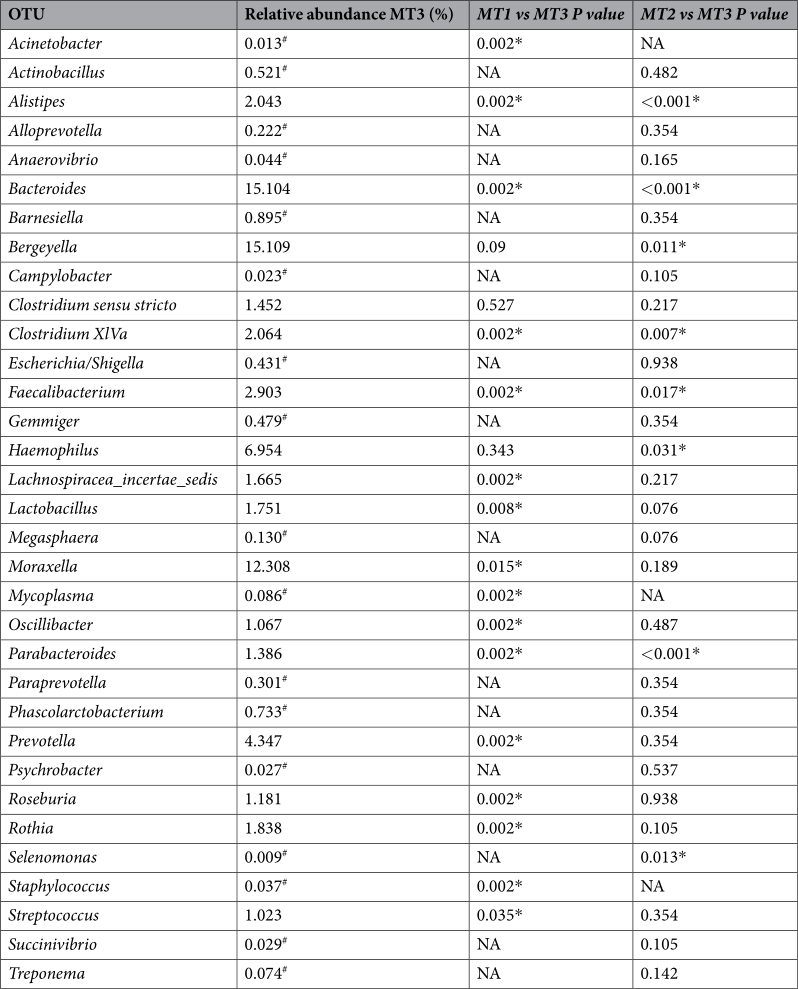
Table 2. Relative abundance of OTUs at third sampling time in MT farm compared with the previous samplings. *Significantly different. # The OTU is included in the list although having relative abundance less than 1% for comparison with previous samplings. NA, not available. When OTUs appeared at less than 1% of relative abundance in both sampling times, the P value was not estimated.
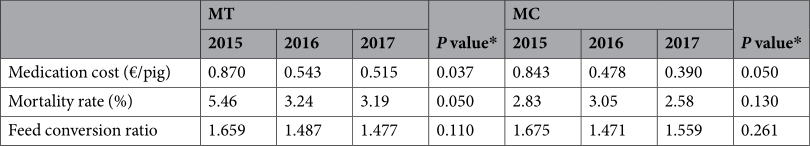
Table 3. Production data collected from the nursery phases of MT and MC farms in three consecutive years. *P values were estimated through Krukal-Wallis non-parametric test.
Material and Methods
Farms and sampling. Nasal swabs from 3-4 week old piglets were collected at different time points, from two different farms (MT and MC) located in the area of Catalonia, Spain. First sampling was done on May-June 2015 when both farms were using perinatal antimicrobial treatments, labelled as MT1 and MC1, for MT and MC farms respectively. MT farm used penicillin and streptomycin at 3 days after birth and tulatromycin one week later; while MC farm used ceftiofur at 3 days and tulatromycin one week later. A second sampling was done after one farrowing interval without usage of any perinatal antimicrobial treatment, in October-November 2015, labelled as MT2 and MC2. In MT farm, a third sampling was performed in March 2017 (labelled as MT3). The MT farm was a 3300-sows multi-site pig production system and MC farm was a 480-sows farrow-to-finish production system. MC farm was negative to porcine reproductive and respiratory syndrome virus (PRRSV) OTU Relative abundance MT3 (%) MT1 vs MT3 P value MT2 vs MT3 P value while MT was PRRS-positive, although no circulation of the virus was diagnosed throughout the study. All the animals were vaccinated against porcine circovirus type 2 (PCV2) and Mycoplasma hyopneumoniae. During the study period no other management practices were modified in any farm, except for the antibiotic treatment elimination. Production data were collected during the period of the study. Collection of nasal swabs (sterile with aluminium handle) was done one day prior weaning from both nostrils (5-6 cm depth) using the same swab. The swabs were kept refrigerated until arrival within 24 hours to the laboratory, where they were stored in PBS (0.5 mL) at -20 °C.
Sampling of piglets was done under institutional authorization and followed good veterinary practices. According to European (Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes) and Spanish (Real Decreto 53/2013) normative, this procedure did not require specific approval by an Ethical Committee. Nasal sampling was performed only once to each piglet and is not likely to cause pain, suffering, distress or lasting harm equivalent to, or higher than, that caused by the introduction of a needle in accordance with good veterinary practice (Chapter I, Article 1, 5 (f) of 2010/63/EU).
DNA extraction and sequencing. Total genomic DNA was extracted using Machinery Nigel Kit (GmbH & Co, Düren; Germany), following manufacter's instructions and resuspended in 50 µl of elution buffer. Quality and quantity was evaluated on a BioDrop DUO (BioDrop Ltd, Cambridge. UK). The library preparation for sequencing was performed within 24 h after the DNA extraction at Servei de Genòmica, Universitat Autònoma de Barcelona. Sequencing of the V3-V4 region of 16S rDNA gene was done with Illumina MiSeq pair-end 2 × 250 bp technology following the manufacturer instructions (MS-102–2003 MiSeq® Reagent Kit v2, 500 cycle). The region targeted to perform the 16S amplification was the one spanning the V3 and V4 region of 16S rRNA gene selected from Klindworth et al. 33 . Interest-specific primers targeting this region were the ones recommended by Illumina with overhang adapters attached:
16S Forward Primer 5' TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG
16S Reverse Primer 5' GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC.
Cycling conditions were the same reported previously 6 . The PCR products were purified and checked to verify its size on a Bioanalyzer DNA 1000 chip (Agilent).
Read filtering and data analysis. For the taxonomic analysis, reads were quality-filtered (Q > 25) before paired-end joining by QIIME v1.9 software 34 , using fastq join 35,36 under default values. Sequences were clustered into OTUs at 97% similarity using UCLUST algorithm 37 and the Greengenes database 38 . Chimeric detection and removal was done with USEARCH 6.1 37,39 against the ChimeraSlayer reference database 40 . To minimize the inflation of rare OTUs in the community analysis, we also exclude singletons for further processing. Taxonomic assignment was done Naïve Bayes classification against RDP database 41 . For each taxon, the Kruskal Wallis test was perform to compare OTU frequencies in sample groups and to ascertain whether or not there are statistically significant differences between the OTU abundance in the different sample groups, P values were FDR-corrected for multiple hypotheses testing. Single rarefaction, based on the sample with the lowest number of reads, was used for alpha-diversity analysis. Diversity indexes were calculated on rarefied 16S rRNA gene sequence data for all samples at 97% similarity. Alpha diversity between groups was compared through two-sample non-parametric t-tests (Monte Carlo method) at maximum depth in rarefied samples (with 999 permutations). In addition, equal number of samples was subsampled to assess the significant differences between sample types using UniFrac weighted and unweighted distances 42,43 . The percentage of variation between grouped samples was measured by R 2 , using adonis function of the vegan package 44 in R software. Estimation of P values was done through Monte Carlo test with 999 random permutations of the data set. Preliminarily, combined data from both farms were compared before and after antimicrobial treatment elimination to explore the data. However, each farm was analysed separately at the different time points throughout all the study. Samples were considered to be significantly different when the accompanying P value was ≤0.05. Results were confirmed with SAS software through PROC GLM analysis.
The production data parameters were statistically analysed through Krukal-Wallis non-parametric test using R software 45 .
For more of the article, please click here.
Article made possible through the contribution of Florencia Correa-Fiz et al.